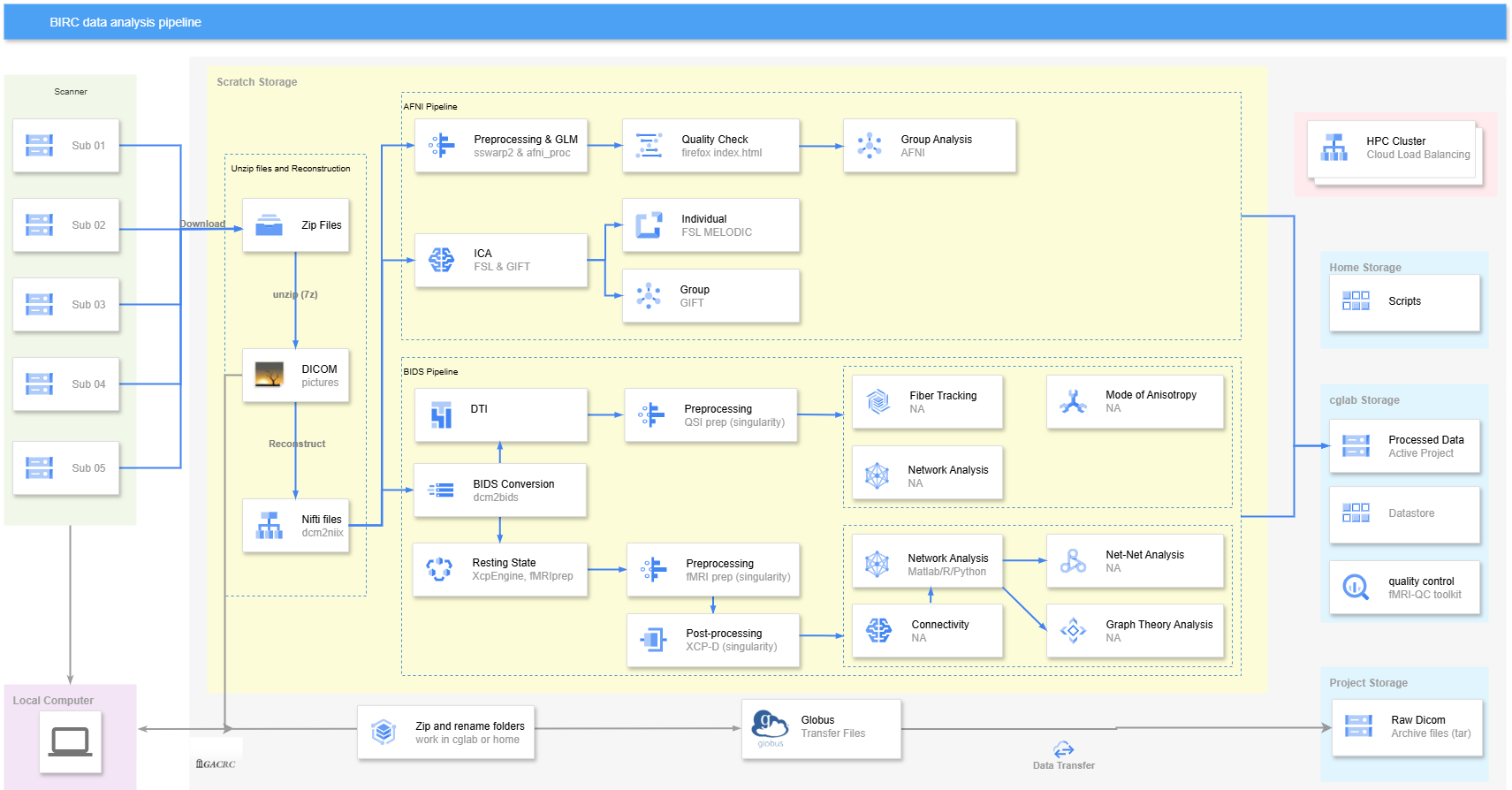
Workflow¶
Analyze multiple subjects at the terminal.
You can use a
for loopto process multiple subjects, or you can open multiple terminals at the same time to run scripts that analyze individual data. However, this approach is demanding on computer resources, takes a long time to run, and does not make good use of HPC.
Analyze single or multiple subjects by submitting jobs.
The syntax for submitting Jobs is not difficult to learn, and you can flexibly adjust the resources of HPC accordingly. The most important thing is that you can submit array jobs to process multiple subjects.
Download Raw Data¶
Go to scratch workspace
cd /scratch/yourIDCreate workDir and rawdata folders.
mkdir workDir/rawdataYou can be very flexible in creating these folders as long as you know how to modify the path in the script.
Load firefox module.
ml FirefoxOpen firefox.
firefoxChange the Downloads path to the folder you just created. Now you can open Outlook by using Firefox you load and start to download the raw data (zip files)!
Unzip¶
Using 7z command will take advantage of parallel tasks. We end up with one (or more) subfolders that contain DICOM images.
First of all, let’s copy the scripts to our rawdata folder. cp /work/cglab/projects/BRANCH/all_data/for_AFNI/processing_scripts/{unzip_all_files,convert_dicom.sh} /scratch/yourID/workDir/rawdata. You can also use GACRAC GUI or Globus to do it.
Load module:
ml p7zip/17.04-GCCcore-11.3.0Unzip files:
7z x download_2024-11-20_20-24-15.zipIf you want to unzip all files in the current folder, use
ml parallelto load parallel module, and then usefind $(pwd) -name "*.zip" | parallel '7z x {}'
Convert the DICOM files to Nifti files¶
The following provides a custom naming scheme, though I recommend converting DICOM to BIDS format. For the theory part, see 02 Reconstruction: Convert 2D images to 3D (anatomical) or 4D (functional)
Use ./convert_dicom.sh in the Terminal to run the script.
The basic logic of this script is to extract the ID from the DICOM file after it is processed in the temporary folder and create the s$ID folder and its subfolders based on the ID. Finally, the corresponding T1w and other files are put into the corresponding folder. In addition, it will automatically copy the script to the appropriate folder.
If you want to read DICOM files use dicom_hdr or dicom_hinfo. For example, sometimes I will want to check if the ID is correct: dicom_hdr i119849059.MRDC.32 | grep "Patient ID"
#!/bin/bash
# Description: reconstruct dicom files with AFNI's dcm2niix_afni program, then categorize files.
# Usage: use it with slurm script.
# Enter working directory
cd "$input_dir" || exit
if [ ! -d "$input_dir" ]; then
echo "Error: $input_dir does not exist, check the path."
exit 1
fi
# create an issue log
log_file="$input_dir/log_$(date '+%Y-%m-%d_%H-%M-%S').txt"
exec > >(tee -a "$log_file") 2>&1
# Iterate through e-prefixed directories
for e_dir in e*/; do
# Remove trailing /
e_dir=${e_dir%/}
# Find first file with MRDC or first file in s-prefixed subdirectories
file=""
for s_dir in "$e_dir"/s*/; do
# Prefer MRDC files, otherwise take first file
file=$(find "$s_dir" -type f | grep -m 1 "MRDC")
# If no MRDC file, take first file
if [ -z "$file" ]; then
file=$(find "$s_dir" -type f -print -quit)
fi
# Exit loop if file found
if [ -n "$file" ]; then
break
fi
done
# Process found file
if [ -n "$file" ]; then
# Extract Patient ID
patient_id=$(dicom_hdr "$file" | grep "Patient ID" | awk -F'//' '{print $3}' | tr -d ' ')
# Extract branch number (remove leading letters and take first number group)
ID=$(echo "$patient_id" | sed -E 's/^[^0-9]*([0-9]+).*/s\1/')
# Create path
subject_dir="$base_dir/$ID"
raw_dir="$subject_dir/raw"
derivatives_dir="$subject_dir/derivatives"
mkdir -p "$raw_dir"
mkdir -p "$derivatives_dir/cards_output" "$derivatives_dir/kidvid_output" "$derivatives_dir/sswarp2" "$derivatives_dir/bias_field_correct" "$derivatives_dir/DTI" "$derivatives_dir/resting_state"
# Convert DICOM files to NIfTI and store in raw directory
dcm2niix_afni -p y -z y -f "%i_%d_%s_%z" -o "$raw_dir" "$e_dir"
# Move files based on naming conventions
find "$raw_dir" -type f | while read file; do
if [[ "$file" == *"T1"* ]]; then # move the T1 files to sswarp2 folder
cp "$file" "$derivatives_dir/sswarp2/"
elif [[ "$file" == *"CARDS"* ]]; then
cp "$file" "$derivatives_dir/cards_output/"
elif [[ "$file" == *"CEV"* ]]; then
cp "$file" "$derivatives_dir/kidvid_output/"
cp "$file" "$derivatives_dir/bias_field_correct/"
elif [[ "$file" == *"DTI"* ]]; then
cp "$file" "$derivatives_dir/DTI/"
elif [[ "$file" == *"RS"* ]]; then
cp "$file" "$derivatives_dir/resting_state/"
elif [[ "$file" == *"PM"* ]]; then
cp "$file" "$derivatives_dir/bias_field_correct/"
fi
done
echo "Finished processing subject $ID" # you will see how afni process each ID
fi
donePreprocessing¶
For the theory part, see 03 Preprocessing & afni_proc.py and 04 General Linear Regression & Experimental Design
Sswarp2¶
We use the sswarper2 command to preprocess T1-weighted (T1w) images (note that this command is only available in AFNI versions from 2024 onward). While afni_proc.py also offers similar preprocessing functionalities, performing separate preprocessing on T1w data beforehand lays a stronger foundation for subsequent functional imaging (fMRI) analysis, as the preprocessed T1w data is generally more precise.
For example, after running
sswarper2, the alignment outputs, such as the standardized T1w image and transformation matrices, can be directly integrated intoafni_proc.py. This stepwise approach offers greater flexibility: if adjustments to alignment or re-evaluation of the T1w data are needed, there is no need to reprocess the entire functional preprocessing pipeline. Additionally, functional data and T1w data can be processed in parallel, saving time.Moreover,
sswarper2employs a specialized skull-stripping algorithm (you don’t need to add extra command in the script) to extract brain tissue more accurately, minimizing the inclusion of non-brain structures such as the face, scalp, or other artifacts.Last,
sswarper2provides a detailed quality control (QC) report that allows for early-stage assessment of data quality. If the T1w data quality is poor, it often indicates that the subsequent fMRI data quality might also be suboptimal (since T1w data reflects structural information and is not influenced by task-related fluctuations). In such cases, we may choose to exclude participants with low-quality data.
Quote from AFNI:
This script has dual purposes for processing a given subject’s anatomical volume:
to skull-strip the brain, and
to calculate the warp to a reference template/standard space. Automatic snapshots of the registration are created, as well, to help the QC process.
This program cordially ties in directly with afni_proc.py, so you can run it beforehand, check the results, and then provide both the skull-stripped volume and the warps to the processing program.
Script and Annotation¶
#!/bin/bash
# --------------------------------------------------------------------------------------
# Description: preprocess structural MRI data with `sswarper2` in AFNI.
# Usage: use it with slurm script.
# --------------------------------------------------------------------------------------
subj="s$1"
t1_dir="$base_dir/$subj/derivatives/sswarp2"
export AFNI_NO_X11=1
# Check if the specified path is a valid directory
if [[ ! -d "$t1_dir" ]]; then
echo "wrong ID"
exit 1
fi
cd "$t1_dir" || exit 1
# Create T1_results folder
mkdir -p "$t1_dir/T1_results"
for nifti_file in BR*.nii.gz; do
if [ -f "$nifti_file" ]; then
echo "processing: $nifti_file"
echo "ID: $subj"
sswarper2 \
-input "$nifti_file" \
-base /work/cglab/projects/BRANCH/all_data/for_AFNI/HaskinsPeds_NL_template1.0_SSW.nii \
-subid "$subj" \
-deoblique_refitly \
-giant_move \
-odir "$t1_dir/T1_results" 2>&1 | tee log_$subj
else
echo "cannot find .nii.gz files"
exit 1
fi
done
echo "finished processing"Annotation:
-input: your input*BRCH*.nii.gzfiles.-base /work/cglab/projects/BRANCH/all_data/for_AFNI/HaskinsPeds_NL_template1.0_SSW.nii: non-linearly warp and transform the participant’s anatomical image to standardized space.-giant_move: remove giant move.-odir: output folder.deoblique_refitly:(opt) purge obliquity information to deoblique the input volume (copy, and then ‘3drefit -deoblique ...’), as an initial step. This might help when data sets are very... oblique.
Output¶
In the T1 results folder, AM*.jpg, MA*.jpg, QC_anatQQ*.jpg and QC_anatSS*.jpg are the images that we care most. In the ssw_aligh_hist.* folder you will see the step-by-step images generated in the process, which is not very necessary.
anatQQ.$ID.nii= skull-stripped dataset nonlinearly warped to the base template space;anatSS.$ID.nii= second pass skull-stripped original dataset; anatS and anatSS are ‘original’ in the sense that they are aligned with the input dataset.The inputs needed for the
-tlrc_NL_warped_dsetsoption to afni_proc.py: not the SS files
-tlrc_NL_warped_dsets \
anatQQ.${subj}.nii \
anatQQ.${subj}.aff12.1D \
anatQQ.${subj}_WARP.niiafni_proc.py¶
There are two versions of regression, A or B. You can find it in papersurvey or other csv files; the file names also tells the version, e.g., BRCH-157-25-123_ECEV_B6.nii.gz is version B.
Script and Annotation¶
#!/bin/bash
# --------------------------------------------------------------------------------------
# Description: AFNI's preprocessing pipeline, for kidvid task, video version A.
#
# Note:
# Remove 6 TRs, the stimulus files must match datasets that have had such TRs removed.
# Some other tips that might be helpful:
# https://afni.nimh.nih.gov/pub/dist/doc/htmldoc/programs/alpha/afni_proc.py_sphx.html#ahelp-afni-proc-py
#
# Usage: use it with slurm script.
# --------------------------------------------------------------------------------------
echo "$1"
subj="s$1"
t1_dir="$base_dir/$subj/derivatives/sswarp2"
cd "${base_dir}/${subj}/derivatives/kidvid_output"
export AFNI_NO_X11=1
afni_proc.py \
-subj_id $subj \
-script proc."${subj}" -scr_overwrite \
-out_dir "${subj}.results" \
-copy_anat "${t1_dir}"/T1_results/anatSS."${subj}".nii \
-anat_has_skull no \
-dsets B*CEV*nii* \
-blocks tshift align tlrc volreg blur mask scale regress \
-radial_correlate_blocks tcat volreg regress \
-tcat_remove_first_trs 6 \
-align_unifize_epi local \
-align_opts_aea -cost lpc+ZZ -giant_move -check_flip \
-tlrc_base /work/cglab/projects/BRANCH/all_data/for_AFNI/HaskinsPeds_NL_template1.0_SSW.nii \
-tlrc_NL_warp \
-tlrc_NL_warped_dsets \
"${t1_dir}"/T1_results/anatQQ."${subj}".nii \
"${t1_dir}"/T1_results/anatQQ."${subj}".aff12.1D \
"${t1_dir}"/T1_results/anatQQ."${subj}"_WARP.nii \
-volreg_align_to MIN_OUTLIER \
-volreg_align_e2a \
-volreg_tlrc_warp \
-volreg_compute_tsnr yes \
-mask_epi_anat yes \
-test_stim_files no \
-blur_size 4.0 \
-regress_stim_times \
"${scripts_dir}"/kidvid_stim_timing_files/A_remove_6TRs/negative.1D \
"${scripts_dir}"/kidvid_stim_timing_files/A_remove_6TRs/neutral.1D \
"${scripts_dir}"/kidvid_stim_timing_files/A_remove_6TRs/positive.1D \
-regress_stim_labels neg neut pos \
-regress_basis_multi 'dmBLOCK' 'dmBLOCK' 'dmBLOCK' \
-regress_stim_types AM1 AM1 AM1 \
-regress_opts_3dD \
-gltsym 'SYM: pos -neut' \
-gltsym 'SYM: neg -neut' \
-gltsym 'SYM: pos -neg' \
-gltsym 'SYM: 0.5*pos +0.5*neg -neut' \
-glt_label 1 Pos-Neut \
-glt_label 2 Neg-Neut \
-glt_label 3 Pos-Neg \
-glt_label 4 PosNeg-Neut \
-regress_censor_motion 0.3 \
-regress_censor_outliers 0.05 \
-regress_motion_per_run \
-regress_3dD_stop \
-regress_reml_exec \
-regress_compute_fitts \
-regress_make_ideal_sum sum_ideal.1D \
-regress_est_blur_epits \
-regress_est_blur_errts \
-regress_run_clustsim no \
-html_review_style pythonic \
-execute \Submit Array Jobs¶
By using array jobs, HPC automatically generates separate parallel jobs for each of your participant, and then runs your main script (the one that processes the individual subjects). Note that AFNI does not benefit from MPI.
You need to change: the range of the array (the total number should be the number of the participants, e.g., 1-2 means 2 participants), which will generate new names for each of your individual jobs; the name of the script to run. After revise the job script, use sbatch script.sh to submit the job.
#!/bin/bash
#SBATCH --job-name=afni_preprocessing # Job name
#SBATCH --partition=batch # Partition (queue) name
#SBATCH --nodes=1
#SBATCH --ntasks=1 # Run a single task
#SBATCH --cpus-per-task=20 # Number of CPU cores per task
#SBATCH --mem-per-cpu=1gb
#SBATCH --time=20:00:00 # Time limit hrs:min:sec
#SBATCH --output=/scratch/%u/workDir/log/preprocessing/afni_%A-%a.out
#SBATCH --error=/scratch/%u/workDir/log/preprocessing/afni_%A-%a.err
#SBATCH --array=1-3 # Array range: DONT FORGET TO CHANGE!!!
# Define base_dir before using it
export myID="$USER"
export base_dir="/scratch/$myID/workDir"
export scripts_dir="/work/cglab/projects/BRANCH/all_data/for_AFNI/processing_scripts"
export my_scripts="/home/$myID/Desktop/scripts"
export AFNI_NO_X11=1
# Load AFNI module
ml Flask/2.3.3-GCCcore-12.3.0
ml netpbm/10.73.43-GCC-12.3.0
ml AFNI/24.3.06-foss-2023a
ml FSL/6.0.7.14-foss-2023a
source ${FSLDIR}/etc/fslconf/fsl.sh
subjects=(198 212 218)
subject=${subjects[$((SLURM_ARRAY_TASK_ID - 1))]}
# Run the scripts
time "$my_scripts/sswarp_scratch_main.sh" ${subject}
time "$my_scripts/bias_field_correction_scratch_v2.0.sh" ${subject}
# A
time "$my_scripts/A_kidvid_preprocessing_add16s.sh" ${subject}
Tar DICOM Files and Transfer to GLOBUS¶
#!/bin/bash
# --------------------------------------------------------------------------------------
# Description:
# Find first file with MRDC or first file in s-prefixed subdirectories in e-prefixed directories,
# extract ID, folder name, etc. and generate a csv file. Rename the e* folders based on this csv file,
# finally, zip all the folders.
#
# Usage: use the script in the terminal.
# --------------------------------------------------------------------------------------
# Load the parallel module
ml parallel/20240322-GCCcore-13.2.0
ml AFNI/24.3.06-foss-2023a
# Set working directory
work_dir="/home/$USER/zip_all_files/"
# Enter working directory
cd "$work_dir" || exit
# Create CSV file
csv_file="dicom_summary_$(date '+%Y-%m-%d_%H-%M-%S').csv"
# Write CSV header
echo "e_Folder Name,ID,Branch_Number,File,File Path" > "$csv_file"
# Iterate through e-prefixed directories
for e_dir in e*/; do
# Remove trailing /
e_dir=${e_dir%/}
# Find first file with MRDC or first file in s-prefixed subdirectories
file=""
for s_dir in "$e_dir"/s*/; do
# Prefer MRDC files, otherwise take first file
file=$(find "$s_dir" -type f | grep -m 1 "MRDC")
# If no MRDC file, take first file
if [ -z "$file" ]; then
file=$(find "$s_dir" -type f -print -quit)
fi
# Exit loop if file found
if [ -n "$file" ]; then
break
fi
done
# Process found file
if [ -n "$file" ]; then
# Extract Patient ID
patient_id=$(dicom_hdr "$file" | grep "Patient ID" | awk -F'//' '{print $3}' | tr -d ' ')
# Extract branch number (remove leading letters and take first number group)
branch_number=$(echo "$patient_id" | sed -E 's/^[^0-9]*([0-9]+).*/s\1/')
# Record in CSV
echo "$e_dir,$patient_id,$branch_number,$(basename "$file"),$file" >> "$csv_file"
fi
done
# Display console output
echo "DICOM Summary:"
echo "Folder Name | Patient ID | Branch Number | First File"
echo "-------------------------------"
cat "$csv_file" | tail -n +2 | column -t -s,
echo -e "\nOutput file created: $csv_file"
# Create a log file for renaming operations
rename_log="rename_log_$(date '+%Y-%m-%d_%H-%M-%S').txt"
echo "Renaming Log - $(date)" > "$rename_log"
# Skip the header and process each line
tail -n +2 "$csv_file" | while IFS=',' read -r folder_name patient_id branch_number first_file file_path
do
# Check if branch_number is valid and different from folder_name
if [ -n "$branch_number" ] && [ "$branch_number" != "$folder_name" ]; then
# Ensure the old directory exists
if [ -d "$folder_name" ]; then
# Attempt to rename
if mv "$folder_name" "$branch_number"; then
echo "Renamed: $folder_name -> $branch_number" >> "$rename_log"
else
echo "Error renaming $folder_name to $branch_number" >> "$rename_log"
fi
else
echo "Directory $folder_name does not exist" >> "$rename_log"
fi
else
echo "Skipping: $folder_name (invalid or same name)" >> "$rename_log"
fi
done
# Display log contents
cat "$rename_log"
# Find and compress each "s" prefixed folder in parallel
find . -maxdepth 1 -type d -name "s*" | parallel -j 10 tar -czf {}.tar.gz {}